Walter Reed Files: I recently had the opportunity to mention Lafora’s Disease in the comment section of my video on adhesive arachnoiditis syndrome and thought it would be a good opportunity to review this disorder. This particular case had a huge impact on me and I grieved for this young lady. To see such a young and vibrant person succumb so rapidly to disease was difficult for me at the time, even though I had years of experience on the medicine wards.
I was no stranger to loss of life from many hours and years in the emergency rooms (from car accidents to gun shot wounds), the medicine wards and intensive care units. I have experienced cancer patients developing tumor erosion into pulmonary arteries…resulting in sudden respiratory bleeding into a bed pan, the patient completely bleeding out within a minute right before my eyes. I remember the patient cycling through severe blood loss, to CNS hypoxia, to full tonic, clonic rigidity and then death. There was nothing I could do. I have no idea how many codes I have performed, but I do remember the meticulous ACLS protocol that I would systematically follow until it’s final conclusion. I have managed two patients with cardiac arrest in the intensive care units at the same time, barking out orders like general Patton…so I was very familiar with death.
These emergency situations are controlled and you move through your management with all the precision you can muster…sometimes you succeed and sometimes you don’t. As Chief Resident at Tripler Army Medical Center I knew what it’s like to have 15 admission in an hour related to real mass casualties that occurred during a training mission on the islands. A smoke bomb deployed in a tunnel during a litter carry exercise turned into a disaster when the smoke did not ventilate properly. Three of these troops, my fellow brothers and sisters in arms, ended up in the intensive care units on ventilators and one of them died. He was so bloody young. I have had amazing experiences in my life of medicine. But at times it takes a toll.
She was 21 years-old and she was admitted for evaluation of myoclonic seizures. At the age of 14 she had her first seizure. She was an “A” student until her sophomore year in college, when she had another generalized seizure. Her academic standing rapidly declined. There was no family history of consanguinity. Her neurologic examination was most notable for severe dementia and frequent small-amplitude myoclonic jerks. Examination of the skin was noncontributory.
An electroencephalogram revealed generalized, high-voltage, irregular decreases in frequency and frequent high-voltage independent spikes, polyspikes, and spike-wave complexes. An electroretinogram was normal. A computerized axial tomogram and a magnetic resonance spectrogram of the head was normal. Lafora’s disease was suspected as the cause.
A biopsy premortem may be obtained from brain, muscle, liver or skin. Carpenter and Karpati were the first to note PAS-positive inclusions in eccrine sweat ducts and subsequent studies had confirmed that random skin biopsies to confirm the diagnosis of Lafora’s disease was a good and less invasive option for confirmation. The diagnosis is established on demonstration of characteristic cytoplasmic inclusions, called Lafora bodies, in the brain, striated muscle, liver or skin. Despite the absence of cutaneous findings, typical periodic acid-Schiff (PAS)-positive inclusions in skin biopsy specimens are present in eccrine ducts, axillary apocrine myoepithelial cells, and peripheral nerves. We published this case to suggest that the axilla should be the site of choice for random cutaneous biopsy to exclude Lafora’s disease because of the presence of both eccrine and apocrine structures in high concentration. Two axillary biopsy specimens in our patient confirmed the diagnosis. To access this article click HERE. Another reference, click HERE.
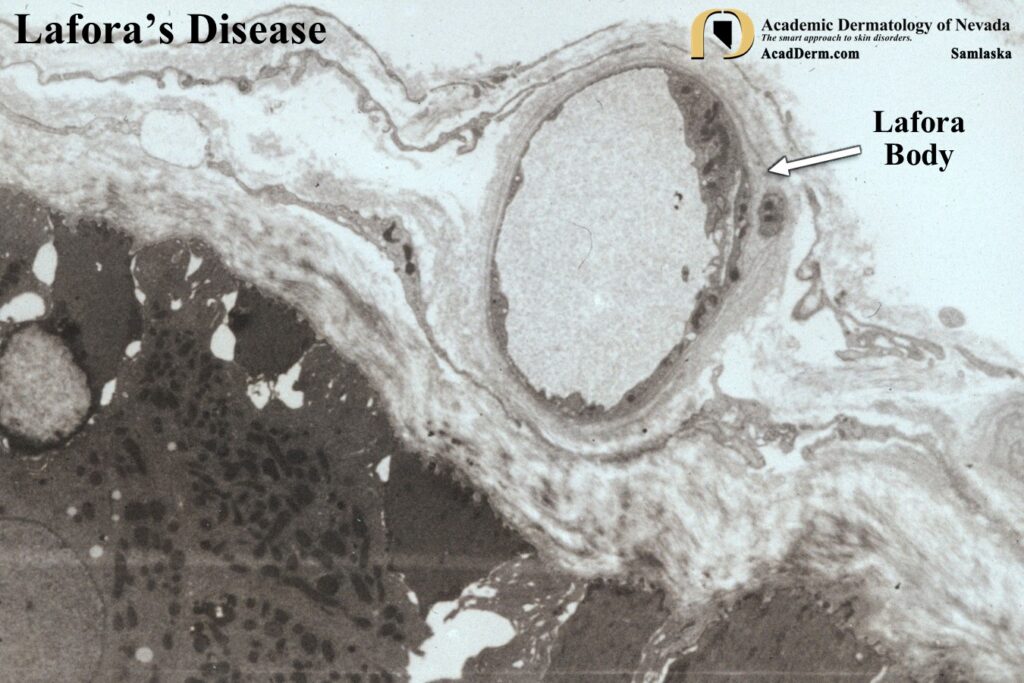
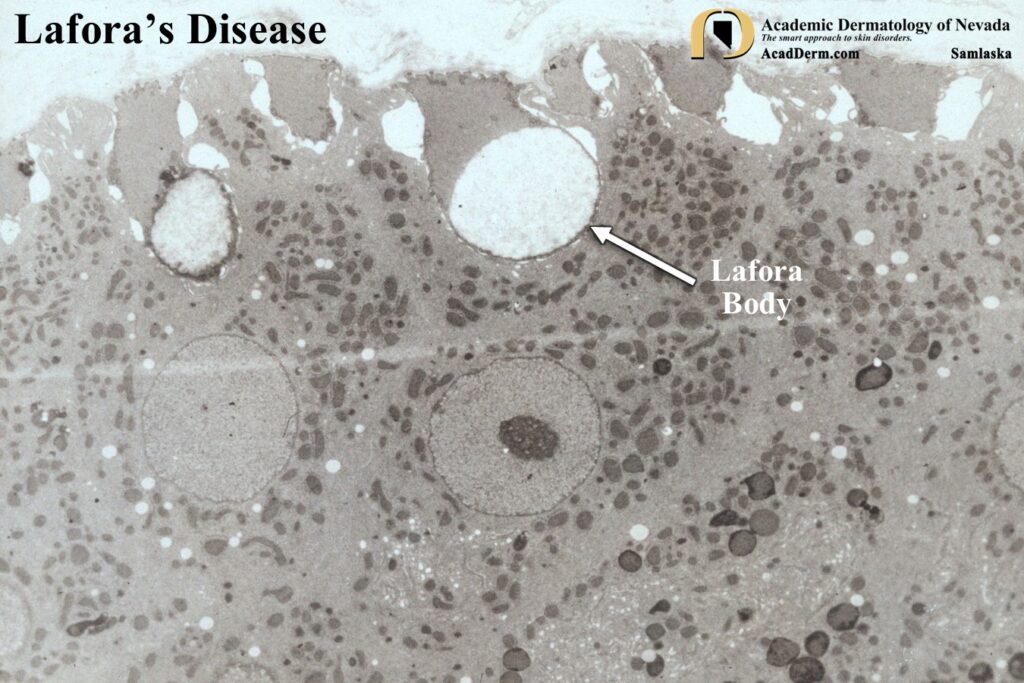
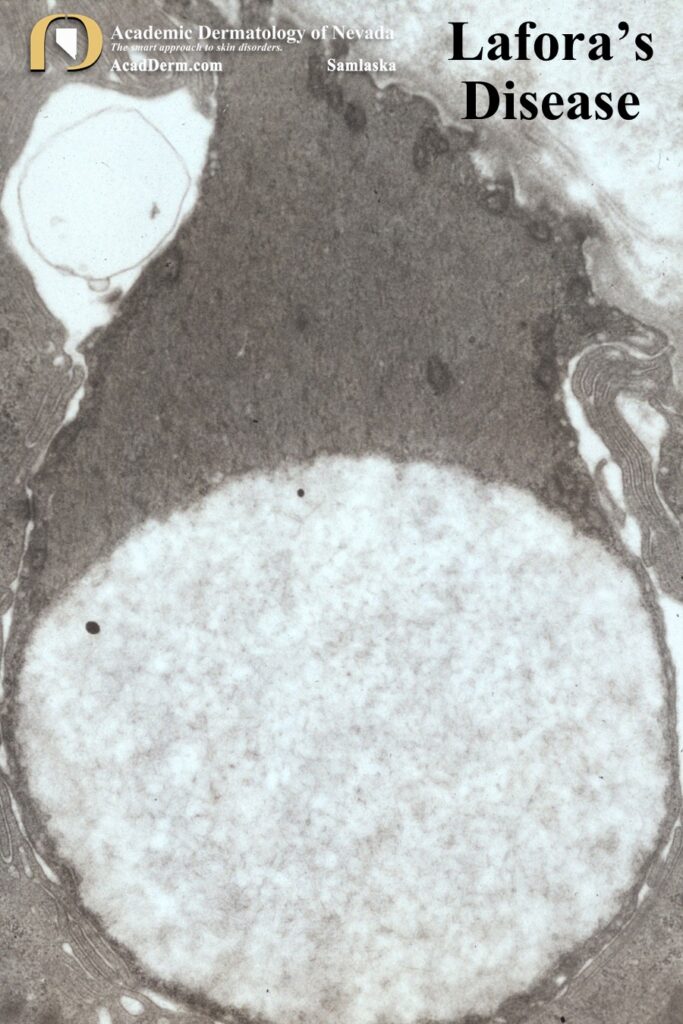
The images above demonstrate Lafora bodies and the filamentous and fine granular matrix within non-membrane-bound inclusion bodies within myoepithelial cells (electron micrograph X 23,700).
What is Lafora’s disease? Lafora’s disease was first described by Univerricht in 1891. The main clinical manifestation are seizures, myoclonus and dementia, with onset occurring between the ages of 10 and 19 years. The initial manifestation are decreased scholastic performance, behavioural disorders, or generalized convulsions. Myoclonic jerks associated with progressive dementia develop within the next few years. Death occurs within 2-10 years as a result of heart failure, liver failure or aspiration pneumonia. The disease is transmitted in an autosomal recessive pattern of inheritance and is frequently associated with parental consanguinity. The diagnosis is confirmed on demonstration of characteristic Lafora bodies at autopsy (in brain, heart, choroid plexus, spinal nerves and striated muscle) or in premortem biopsy specimens of brain, muscle, liver or skin.
ETIOLOGY: Lafora disease is caused by loss-of-function mutations in EPM2A (chromosome 6q24.3) or EPM2B (chromosome 6p22.3) also known as HNLRC1, which encode laforin and malin, respectively. The absence of either protein results in poorly branched, hyperphosphorylated glycogen, which precipitates, aggregates and accumulates into Lafora bodies. It is believed that accumulation of these intracellular inclusions are a principal driver of neurodegeneration and neurological disease. Laforin-malin as an interacting complex suggests that laforin recruits malin to parts of glycogen molecules where overly long glucose chains are forming, so as to counteract further chain extension. In the absence of either laforin or malin function it is believed that long glucose chains in specific glycogen molecules extrude water, form double helices and drive precipitation of those molecules, which over time accumulate into Lafora bodies.
TREATMENT: Needless to say we know much more about Lafora’s disease today then when I published my article. Virus-mediated gene replacement therapy is an exciting development designed to deliver a functional copy of the defective gene. Other delivery methods using nanoparticles, liposomes and exosomes have been suggested. To read further on Lafora’s disease click HERE, HERE and HERE.